


Preventing Cystic Fibrosis
Introduction to Cystic Fibrosis
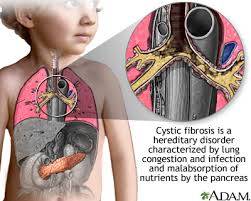
Cystic fibrosis (CF) is a genetic disorder that primarily affects the lungs and digestive system. It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which regulates the flow of salt and fluids in and out of cells. This malfunction leads to the production of thick, sticky mucus that clogs the airways and traps bacteria, causing recurrent lung infections and progressive damage to the respiratory system. Additionally, CF can impair the function of the pancreas, leading to digestive problems and malnutrition.
While cystic fibrosis is a lifelong condition that currently has no cure, advancements in medical research and treatments have significantly improved the quality of life and life expectancy for individuals with CF. However, prevention and early intervention remain crucial in managing the disease and minimizing its impact on affected individuals and their families.

Understanding the Genetics of Cystic Fibrosis
Cystic fibrosis is an autosomal recessive disorder, meaning that an individual must inherit two copies of the defective CFTR gene (one from each parent) to develop the disease. If both parents are carriers of the CF gene mutation, there is a 25% chance with each pregnancy that their child will inherit two copies of the mutated gene and develop cystic fibrosis, a 50% chance that the child will be a carrier like the parents, and a 25% chance that the child will neither have CF nor be a carrier.
Genetic testing can identify carriers of the CF gene mutation, allowing individuals to make informed decisions about family planning and providing valuable information for genetic counseling. Prenatal screening and genetic counseling can help prospective parents understand their risk of having a child with cystic fibrosis and explore options such as in vitro fertilization (IVF) with preimplantation genetic diagnosis (PGD) to select embryos without the CF gene mutation.
Preventing Cystic Fibrosis Through Genetic Screening and Counseling
Genetic screening for cystic fibrosis is recommended for individuals or couples with a family history of CF or those belonging to high-risk ethnic groups, such as Caucasians of Northern European descent, where the prevalence of CF mutations is higher. Carrier screening can be performed using blood tests or saliva samples to identify individuals who carry one copy of the CF gene mutation.
Genetic counseling plays a vital role in helping individuals understand their carrier status, assess their risk of having a child with cystic fibrosis, and explore available reproductive options. Couples who are both carriers of the CF gene mutation may choose to undergo prenatal testing during pregnancy to determine if the fetus has inherited CF and make informed decisions about the continuation of the pregnancy.
Advances in Assisted Reproductive Technologies (ART) have expanded options for couples at risk of having a child with cystic fibrosis. In vitro fertilization (IVF) with preimplantation genetic diagnosis (PGD) allows embryos to be screened for the CF gene mutation before implantation, enabling couples to select unaffected embryos for transfer and reduce the risk of passing on the disease to their children.
Early Detection and Diagnosis of Cystic Fibrosis
Early detection and diagnosis of cystic fibrosis are essential for initiating appropriate medical interventions and therapies to manage the symptoms and complications of the disease. Newborn screening programs, which involve testing infants shortly after birth for genetic conditions like cystic fibrosis, have become standard practice in many countries, enabling early detection and intervention.
Newborn screening for CF typically involves collecting a small blood sample from the infant's heel and analyzing it for elevated levels of immunoreactive trypsinogen (IRT), a protein that is often elevated in newborns with CF. Infants who screen positive for elevated IRT levels undergo further testing, such as a sweat test or genetic testing, to confirm the diagnosis of cystic fibrosis.
The sweat test is considered the gold standard for diagnosing cystic fibrosis and involves measuring the concentration of chloride in a sweat sample collected from the skin. Individuals with CF have abnormally high levels of chloride in their sweat due to the dysfunctional CFTR protein, confirming the diagnosis of the disease.
Treatment and Management Strategies for Cystic Fibrosis
While there is currently no cure for cystic fibrosis, advances in medical research and treatment have significantly improved the prognosis and quality of life for individuals living with the disease. The goals of treatment for CF are to alleviate symptoms, prevent complications, and improve overall health and well-being.
Multidisciplinary care teams, consisting of pulmonologists, gastroenterologists, nurses, dietitians, respiratory therapists, and social workers, collaborate to provide comprehensive care for individuals with cystic fibrosis. Treatment plans are tailored to each patient's specific needs and may include a combination of the following strategies:
1. Airway Clearance Techniques: Regular airway clearance techniques, such as chest physiotherapy, percussion, vibration, and postural drainage, help loosen and remove mucus from the lungs, improving breathing and reducing the risk of respiratory infections.
2. Medications: Various medications are used to manage the symptoms and complications of cystic fibrosis. Bronchodilators help open the airways and improve airflow, while mucolytics thin mucus secretions, making them easier to clear from the airways. Antibiotics are used to treat and prevent lung infections, and anti-inflammatory drugs help reduce airway inflammation.
3. Nutritional Support: Individuals with cystic fibrosis may have difficulty digesting and absorbing nutrients from food due to pancreatic insufficiency. Nutritional supplementation, pancreatic enzyme replacement therapy, and dietary modifications are used to optimize nutrition and prevent malnutrition.
4. Exercise and Physical Activity: Regular exercise and physical activity are important for maintaining lung function, improving cardiovascular health, and enhancing overall fitness and well-being in individuals with cystic fibrosis.
5. Lung Transplantation: In severe cases of cystic fibrosis where lung function becomes severely compromised despite medical treatment, lung transplantation may be considered as a life-saving intervention. Lung transplantation can improve survival and quality of life in select patients with end-stage lung disease due to CF.
In addition to medical interventions, lifestyle modifications, such as avoiding tobacco smoke, maintaining a healthy diet, staying hydrated, and adhering to prescribed treatment regimens, play a critical role in managing cystic fibrosis and minimizing disease progression.
Promising Advances in Cystic Fibrosis Research
Ongoing research efforts continue to explore new treatment modalities and therapeutic approaches for cystic fibrosis, with the aim of improving outcomes and quality of life for individuals living with the disease. Some promising areas of research include:
1. Gene Therapy: Gene therapy aims to correct the underlying genetic defect responsible for cystic fibrosis by delivering functional copies of the CFTR gene to affected cells. While gene therapy approaches are still in the experimental stages, they hold the potential to provide a long-term cure for CF by addressing the root cause of the disease.
2. CFTR Modulator Therapies: CFTR modulator therapies are a new class of medications that target the defective CFTR protein and improve its function. These medications, such as ivacaftor, lumacaftor, tezacaftor, and elexacaftor, have shown promising results in clinical trials, leading to improvements in lung function, respiratory symptoms, and overall health in individuals with specific CFTR mutations.
3. Personalized Medicine: Advances in genomic medicine and precision medicine have paved the way for personalized treatment approaches tailored to the individual genetic
profile of each patient with cystic fibrosis. By identifying specific CFTR mutations and targeting treatments accordingly, personalized medicine holds the potential to optimize therapeutic outcomes and minimize adverse effects.
4. Novel Therapeutic Targets: Researchers are exploring novel therapeutic targets and treatment strategies for cystic fibrosis, including anti-inflammatory agents, antimicrobial peptides, and modulators of mucin production and secretion. These emerging therapies aim to address key aspects of CF pathophysiology and improve clinical outcomes for affected individuals.

5. Biomarkers and Disease Monitoring: Biomarkers play a crucial role in disease monitoring and treatment response assessment in individuals with cystic fibrosis. Advances in biomarker discovery and validation enable early detection of disease progression, prediction of clinical outcomes, and optimization of treatment strategies, leading to improved patient care and management.
Conclusion
Cystic fibrosis is a complex genetic disorder that affects multiple organ systems, primarily the lungs and digestive system. While there is currently no cure for CF, advances in medical research and treatment have revolutionized the management of the disease, significantly improving the quality of life and life expectancy for affected individuals.
Prevention and early intervention are key components of cystic fibrosis management, involving genetic screening, prenatal testing, newborn screening, and genetic counseling to identify at-risk individuals, facilitate early detection and diagnosis, and explore available treatment options. Multidisciplinary care teams collaborate to provide comprehensive care for individuals with CF, incorporating a range of medical, nutritional, and supportive interventions tailored to each patient's specific needs.
Ongoing research efforts continue to advance our understanding of cystic fibrosis pathophysiology and identify new treatment modalities and therapeutic targets. Gene therapy, CFTR modulator therapies, personalized medicine, and novel therapeutic approaches offer promising avenues for improving outcomes and quality of life for individuals living with cystic fibrosis.
By raising awareness, promoting genetic screening, supporting research initiatives, and advocating for access to comprehensive care and innovative treatments, we can work together to prevent and effectively manage cystic fibrosis, ultimately improving the lives of those affected by this challenging disease.
No comments yet
Be the first to share your thoughts!