


Thalassemia: Causes, Symptoms, Diagnosis, and Treatment
Introduction to Thalassemia
Thalassemia is a group of genetic blood disorders characterized by abnormal hemoglobin production, leading to ineffective erythropoiesis (the process of red blood cell formation) and anemia. It is one of the most prevalent genetic disorders globally, particularly in populations with a high prevalence of carriers, such as those of Mediterranean, Middle Eastern, Southeast Asian, and African descent. Thalassemia poses significant health challenges, but advancements in medical science have improved the quality of life and life expectancy for affected individuals.
This comprehensive guide aims to shed light on the various aspects of thalassemia, including its causes, symptoms, diagnosis, and treatment options.

Understanding Hemoglobin and Its Role
To comprehend thalassemia, it's crucial to understand the role of hemoglobin in the body. Hemoglobin is a protein found in red blood cells that carries oxygen from the lungs to tissues throughout the body and returns carbon dioxide from the tissues back to the lungs for exhalation. It consists of four protein chains: two alpha-globin chains and two beta-globin chains.
In thalassemia, there is a defect in the production of one or more of these globin chains. This results in abnormal hemoglobin production, leading to various health complications.
Types of Thalassemia
Thalassemia is classified into two main types based on the globin chain affected: alpha thalassemia and beta thalassemia.
1. Alpha Thalassemia: In alpha thalassemia, there is a reduction or absence of alpha-globin chain production. The severity of the condition depends on the number of affected genes. The types of alpha thalassemia include:
- Alpha thalassemia minor: Two out of four alpha-globin genes are affected.
- Hemoglobin H disease: Three out of four alpha-globin genes are affected.
- Hydrops fetalis: All four alpha-globin genes are affected, leading to a severe form of thalassemia that is often fatal before or shortly after birth.
2. Beta Thalassemia: In beta thalassemia, there is a reduction or absence of beta-globin chain production. The severity of the condition varies depending on the extent of the deficiency. The types of beta thalassemia include:
- Beta thalassemia minor: One beta-globin gene is affected, resulting in mild symptoms.
- Beta thalassemia intermedia: Both beta-globin genes are affected, leading to moderate to severe symptoms.
- Beta thalassemia major (also known as Cooley's anemia): Both beta-globin genes are severely affected, resulting in a life-threatening condition that requires regular blood transfusions and medical intervention.
Causes of Thalassemia
Thalassemia is primarily caused by genetic mutations that affect the production of alpha or beta-globin chains. These mutations can be inherited from one or both parents who carry thalassemia genes. The inheritance pattern varies depending on the type of thalassemia:
- Alpha Thalassemia: The inheritance pattern of alpha thalassemia can be complex due to the presence of four alpha-globin genes. Depending on which genes are affected and how many are affected, individuals can inherit different forms of alpha thalassemia.
- Beta Thalassemia: Beta thalassemia follows an autosomal recessive inheritance pattern. This means that both parents must carry a mutated beta-globin gene for their child to inherit beta thalassemia. If both parents are carriers, there is a 25% chance with each pregnancy that their child will have beta thalassemia major.
Symptoms of Thalassemia
The symptoms of thalassemia vary depending on the type and severity of the condition. Individuals with thalassemia may experience the following symptoms:
- Fatigue and weakness due to anemia
- Pale or yellowish skin (jaundice)
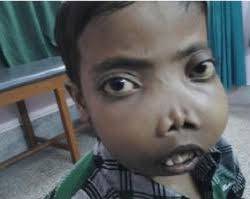
- Bone deformities, especially in the face and skull (in severe cases)
- Delayed growth and development in children
- Enlarged spleen and liver
- Dark urine due to increased breakdown of red blood cells
- Shortness of breath
- Leg ulcers
- Poor appetite
It's essential to note that individuals with thalassemia may not exhibit symptoms until later in childhood or adolescence, especially in cases of alpha thalassemia minor or beta thalassemia minor.
Diagnosis of Thalassemia
Diagnosing thalassemia involves several steps, including:
1. Physical Examination: A healthcare provider may conduct a physical examination to assess symptoms such as pallor, enlarged spleen, or jaundice.
2. Blood Tests: Blood tests are crucial for diagnosing thalassemia. These tests include:
- Complete Blood Count (CBC): This test measures the number of red blood cells, white blood cells, and platelets in the blood. Individuals with thalassemia often have low levels of red blood cells and hemoglobin.
- Hemoglobin Electrophoresis: This test identifies the types of hemoglobin present in the blood and helps differentiate between thalassemia and other types of anemia.
- Genetic Testing: Genetic testing can confirm the presence of specific gene mutations associated with thalassemia.
3. Prenatal Testing: Prenatal testing, such as chorionic villus sampling (CVS) or amniocentesis, may be recommended for pregnant women with a family history of thalassemia to determine if the fetus is affected.
Treatment Options for Thalassemia
The treatment approach for thalassemia depends on the type and severity of the condition. Treatment goals typically include managing symptoms, preventing complications, and improving quality of life. Treatment options may include:
1. Blood Transfusions: Individuals with severe forms of thalassemia, such as beta thalassemia major, require regular blood transfusions to maintain adequate hemoglobin levels and alleviate anemia-related symptoms. However, frequent blood transfusions can lead to iron overload, which requires management with chelation therapy.
2. Chelation Therapy: Chelation therapy involves the use of medications, such as deferoxamine or deferasirox, to remove excess iron from the body. Iron overload can occur due to the regular transfusion of red blood cells, and if left untreated, it can lead to organ damage and other complications.
3. Bone Marrow Transplantation: Bone marrow transplantation, also known as hematopoietic stem cell transplantation, is a potential cure for thalassemia. This procedure involves replacing defective bone marrow with healthy stem cells from a compatible donor. However, bone marrow transplantation carries risks and may not be suitable for all individuals with thalassemia.
4. Gene Therapy: Gene therapy is an emerging treatment approach for thalassemia that involves modifying or replacing defective genes responsible for the condition. While still in the experimental stages, gene therapy shows promise as a potential cure for thalassemia in the future.
5. Supportive Care
: Supportive care measures, such as folic acid supplementation, regular monitoring of iron levels, and management of complications, play a crucial role in improving the quality of life for individuals with thalassemia.
Living with Thalassemia
Living with thalassemia can present various challenges, both physical and emotional. However, with proper medical care, support, and adherence to treatment recommendations, individuals with thalassemia can lead fulfilling lives. It's essential for individuals with thalassemia to:

- Follow a treatment plan outlined by their healthcare provider, which may include regular blood transfusions, chelation therapy, and other supportive measures.
- Stay up-to-date with medical appointments and monitoring tests to detect and manage complications early.
- Maintain a healthy lifestyle, including a balanced diet, regular exercise, and avoiding factors that can exacerbate symptoms or complications, such as smoking or excessive alcohol consumption.
- Seek support from healthcare professionals, support groups, or mental health professionals to address any emotional or psychological challenges associated with living with a chronic condition.
Conclusion
Thalassemia is a complex genetic blood disorder that requires comprehensive management and care. While there is no cure for thalassemia, advancements in medical science have significantly improved treatment options and outcomes for affected individuals. Through early diagnosis, personalized treatment plans, and ongoing support, individuals with thalassemia can live fulfilling lives and effectively manage their condition. Continued research and innovation in thalassemia treatment hold promise for further improving the quality of life and prognosis for individuals affected by this disorder.
No comments yet
Be the first to share your thoughts!